Конференции
68-я Всероссийская научная конференция МФТИ
Список разделов ФЭФМ - Секция квантовой и вычислительной химии
На секцию будут приниматься доклады по исследованию электронной структуры, потенциальных поверхностей, спектральных параметров, термохимических данных, реакционной способности и динамике превращений молекул. Работы по разработке новых методов, алгоритмов и программ расчета волновых функций, строения, свойств и превращений молекул на основе представлений квантовой теории и методов вычислительной математики.
Контакты:mitin.av@mipt.ru
Формат проведения: онлайн
Дата и время проведения:31.03.2026 в 10:00
Место проведения: онлайн платформа
-

Методом цилиндрических волн рассчитаны электронные, спиновые и транспортные свойства хиральных цепочек Te. Оценки спиновой проводимости получены в предположении, что в переносе спина и участвуют преимущественно электроны граничных спиновых состояний, и что вероятность туннелирования электронов выше при параллельной ориентации спина и хиральности материала. Выбором цепочек разной хиральности, можно обеспечить перенос электронов с противоположными спинами в противоположных направлениях
-

Для неконтрактированной многоконфигурационной теории возмущений с использованием потенциала ионизации и сродства к электрону CDAS-PT2-IPEA выведена и реализована поправка первого порядка к дипольному моменту.
-

Представлена реализация метода ACBN0 для расчета поправки Хаббарда в программный пакет FHI-aims. Тесты показали улучшение предсказания ширины запрещенной зоны по сравнению с функционалами SCAN и PBE, сравнимое с гибридным функционалом HSE06. Проектор Левдина обеспечивает стабильность для базиса любого размера. Для перовскитов Ca₂CoNbO₆ и Sr₂SmNbO₆ продемонстрировано корректное описание магнитных состояний. Подход пригоден для исследования коррелированных систем, поверхностей и кластеров.
-
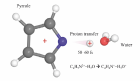
Методом EOM-DIP-CCSD квантохимического пакета Q-chem была проведено молекулярнодинамическое моделировние миграции протона в дикатионных состояниях системы пиррол-вода с использованием разработанного программного интерфейса к платформе для молекулярной динамики Newton-X. Было оценено время миграции протона для различных электронных состояний.
-

В работе рассматривается новый подход к поиску высокоэнергетических молекул посредством машинного обучения. В качестве основы для обучения применяется комбинация структурных дескрипторов языка ФКСП (фрагментарный код суперпозиции подструктур) и квантовые индексы полученные через вычислительные модели. Предполагается, что такой подход поможет извлечь из обучающей выборки локальные теории наличия или отстуствия целевого свойства.
-
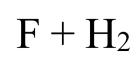
Поуровневые константы скорости, потребность в которых возникает в некоторых задачах астрохимии, получены ab initio для реакции фтора с молекулярным водородом и применены в простой астрохимической модели.
-

В настоящее время международной коллаборацией учёных на базе Массачусетского технологического института ставится эксперимент по измерению эффектов, нарушающих пространственную четность в катионе 29Si16O+. Интерпретация экспериментальных результатов требует теоретического описания молекулярной структуры изучаемой системы. В настоящей работе акцентировано внимание на решении задачи связанных колебательных каналов, а именно — на реализации методов ее решения и их применении.
-
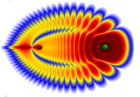
Мы извлекли эффективные квантовые числа ν(R) и функции квантового дефекта μ(R) из прецизионных ab initio потенциалов и применили их для аналитического описания неадиабатических матричных элементов, что обеспечило высокоточное воспроизведение экспериментальных колебательных уровней триплетных состояний H₂ и ее изотопологов.
-

Методом DFT с функционалами B3LYP, PW91, PBE изучается проникновение атомов и ионов бора и бериллия внутрь фуллерена C60 по четырем наиболее характерным направлениям (через центр 6- и 5-угольного кольца, середины двойной и одинарной связей). Построены графики энергии системы в зависимости от положения проникающего атома, получены высота барьера, геометрия переходных состояний, оценка избыточной кинетической энергии атома, обусловленной вовлечением в движение атомов фуллерена.
-

В работе проведено исследование кристаллической структуры тетрафторида тория. Построены две кластерные модели с CTEP. Для них проведен анализ структурных параметров и определена ширина запрещенной зоны в рамках приближения «HOMO-LUMO». Показана необходимость выполнения более точных расчетов фрагментов кристалла с использованием методов теории волновой функции и прецизионных вариантов псевдопотенциалов, поэтому в настоящее время проводятся пилотные расчеты методом связанных кластеров.
-

В данной работе выполнено систематическое первопринципное исследование кластеров системы B–O в широком диапазоне составов BₙOₘ (0 ≤ n, m ≤ 12).
-

Работа посвящена теоретическому исследованию атомов в сильных магнитных полях, характерных для белых карликов и нейтронных звёзд. -

Предложен метод расчёта электронных переходов в соединениях лантанидов на основе квазирелятивистской многочастичной теории возмущений. Показано, что расширение модельного пространства за счёт отбора наиболее значимых детерминантов позволяет снизить погрешность расчёта энергий f–d-переходов до менее 0.1 эВ, сохраняя вычислительную осуществимость метода. Метод успешно протестирован на ионах Ce3+, Pr3+ и кластерной модели твердого тела кристалла ксенотима (YPO4) с примесным центром Ce3+.