68-я Всероссийская научная конференция МФТИ
Список разделов ЛФИ - Секция современных методов атомистического моделирования в науках о материалах
Секция посвящена вопросам предсказания свойств материалов методами атомистического моделирования и искусственного интеллекта.
Основные направления:
-атомистическое моделирование с машинно-обучаемыми потенциалами
-поиск и предсказание свойств новых материалов методами ИИ
Контакты: nikita.orekhov@phystech.edu
Формат проведения: смешанный
Дата и время проведения: 31.03.2026 в 10:00
Место проведения: МФТИ, 2.35 Цифра
-

Работа посвящена применению методов машинного обучения для оптимизации планирования вычислительных протоколов в вычислительном материаловедении. Показано, что подход снижает вычислительные затраты и ускоряет получение результатов.
-

Разработана методология построения фазовых диаграмм тернарных систем на основе первопринципных расчетов, молекулярной динамики и байесовского машинного обучения. Свободные энергии Гиббса восстанавливаются по данным молекулярнодинамических симуляций со статистической неопределенностью с использованием гауссовской регрессии, что позволяет строить фазовые границы с доверительными интервалами. Исследованы бинарные системы In–Au и In–Hg и тернарная система In–Au–Hg.
-

Метод построения бинарных фазовых диаграмм на основе байесовского подхода был обобщён для тернарного случая. Для системы фторидов LiF-NaF-KF был обучен MTP-потенциал и набраны данные из молекулярной динамики. С помощью регрессии гауссовских процессов восстановлена свободная энергия со статистической неопределенностью и построена фазовая диаграмма во всём диапазоне T-c. Результаты хорошо согласуются с данным эксперимента. Метод показал высокую эффективность: затраты составили ~ 100 000 CPU-часов.
-

В работе представлена методика поиска нелинейно-оптических материалов с высокой генерацией второй гармоники при помощи нейросети OptiXNet. Важным достижением стало расширение скрининга на новые диапазоны и способность алгоритма предсказывать свойства полуметаллов, где стандартные квантовые расчеты часто неэффективны. В результате анализа модель открыла ряд новых перспективных соединений для современной оптоэлектроники и фотоники.
-

В работе предложен метод оценки пористости поверхности никелида титана с применением оптической когерентной томографии (ОКТ) и методов машинного обучения. На базе метода Random Forest Regression была создана предиктивная модель, прогнозирующая температуру спекания, непрямую меру пористости, по пространственным статистикам 1 и 2 рода карт глубин, рассчитанных на основе ОКТ-сканов. Коэффициент детерминации R2 данной модели равен 0.95, что указывает на её высокую прогностическую точность.
-

В работе представлен детальный анализ экспериментальных исследований по фоторасслоению As2S3. Так, впервые представлен протокол построения направлений межслоевых сил для двумерных материалов по результатам первопринципных расчётов с решением GW@BSE и анализом частичных волновых функций с локализацией максимального перехода в обратном пространстве ячейки. Разработанный протокол был валидирован на материалах MoS2, WS2, гетероплёнках MoS2/WS2, MoSe2/WSe2 и успешно применён к As2S3.
-

В работе продемонстрировано, как генеративные модели, обученные на больших массивах кристаллических данных, могут быть использованы для целенаправленного поиска материалов с высокой оптической анизотропией. Предложенный подход объединяет генерацию материалов с помощью диффузионных моделей, их эквивариантные графовые представления и DFT-валидацию в единую итеративную систему ускоренного поиска новых оптических кристаллов.
-
Изучаются динамические свойства точечных дефектов в алмазе. Наиболее часто встречающийся примесный атом - азот, оптические центры, в состав которых он входит, имеют прикладные свойства для создания квантовых компьютеров и нанометок в технологии промышленной трассировке алмазов. Работа выполнена методом молекулярной динамики с использованием машинно-обучаемых потенциалов. Вычислены энергии активации и коэффициенты диффузии различных дефектов.
-

В работе проведено теоретическое исследование влияния дефектов и допирования азотом углеродного носителя на свойства наночастицы высокоэнтропийного сплава PtPdCuNiCo с использованием метода теории функционала плотности. Показано, что наличие дефектов в структуре графена способствует увеличению переноса электронов на адсорбированную молекулу кислорода, что облегчает её диссоциацию.
-

Работа фокусируется на проведении DFT-расчетов VASP для поиска высокоанизотропных оптических материалов в генеративных фреймворках. Исследуются методы повышения производительности расчетов и пропускной способности для валидации сгенерированных структур.
-

Исследованы процессы имплантации серы в кремний и последующего термического отжига при высоких уровнях допирования. Моделирование выполнено в рамках молекулярной динамики с использованием нейроэволюционного машинно-обучаемого потенциала NEP, обученного на данных теории функционала плотности.
-

В работе проведено теоретическое исследование механических свойств моно- и поликристаллического высокоэнтропийного диборида при растяжении и изгибе. Моделирование выполнено в рамках молекулярной динамики с использованием машинно-обучаемого потенциала DeepMD. Вычислены модули Юнга для различных и показано, что в поликристаллических образцах наблюдается инверсный эффект Холла-Петча, приводящий к снижению жесткости материала при уменьшении размера зерна.
-

Моделирование термодинамических свойств диамана с применением методов машинного обучения
-

Прогнозирование энергии адсорбции на металлических наночастицах, особенно высокоэнтропийных сплавах, затруднено сложной атомной структурой и ограниченностью простых дескрипторов. В работе применены методы машинного обучения для построения дескрипторов на примере наночастиц AgAuCuIrPdPtRhRu при адсорбции NO и CO. Результаты подтверждают нелинейную зависимость энергии адсорбции от свойств поверхности, а метод SISSO позволяет получить компактные аналитические выражения для поиска новых материалов.
-

В работе предложен автоматизированный метод расчета термодинамических свойств твердых и жидких фаз материалов на основе байесовского восстановления свободной энергии по данным молекулярной динамики. Подход позволяет предсказывать свойства с доверительными интервалами и включает квантовую коррекцию для описания низкотемпературной области. Метод реализован в ансамблях NVT и NPT и применяется во всем диапазоне температур, включая расчет свойств в точке плавления.
-
Данное исследование даёт исчерпывающее представление об адсорбционных и каталитических свойствах поверхностей высокоэнтропийного карбида (HfTaZrNbTiC5), а также о возможностях его применения для различных каталитических процессов. Также в ходе настоящего исследования были получены данные о влиянии атомного окружения на адсорбционные свойства каждого из металлов, входящих в состав HfTaZrNbTiC5, что является важным фундаментальным знанием о высокоэнтропийных соединениях.
-

В работе представлены первые шаги в исследовании особенностей физико-химических свойств на границе раздела между УНТ и стержневой структурой Te. В ходе работы были проведены моделирование и оценка стабильности структур, состоящих из одностенных углеродных нанотрубок разных диаметров (8 – 11 Å) и хиральностей с размещенными внутри стержнями теллура.
-

В данной работе рассмотрен подход, реализующий активное обучение на локальных атомных окружениях, для моделирования многокомпонентных систем.
-

Целью данной работы является атомистическое исследование образования и эволюции цепочечных фрагментов в BN-нанотрубках при одноосном растяжении в широком диапазоне температур и диаметров. Для анализа структуры выполняется автоматическое выделение связей B–N и анализ графа, позволяющий определять цепочечные фрагменты, их длины и время жизни. Планируется построить зависимость образования цепочек от температуры и диаметра нанотрубок, распределения их длин и характерных времён жизни.
-

В работе первопринципными методами исследованы адсорбционные и каталитические свойства наночастиц борида вольфрама в реакции разложения аммиака как безуглеродного способа получения водорода. Показано, что NH₃ молекулярно адсорбируется на W-сайтах с энергиями, сопоставимыми с традиционными металлическими катализаторами, при умеренной стабилизации атомарного водорода. Полученный баланс адсорбционных свойств указывает на перспективность W-B наночастиц для катализа разложения аммиака.
-

В работе методами DFT, NEB и ab initio молекулярной динамики исследованы дефекты и миграция Li⁺ в объёмно-центрированном кубическом Li и твёрдых растворах Li₁₋ₓMₓ. Рассчитаны энергии образования дефектов, связи «вакансия–допант», энергетические барьеры миграции, коэффициенты диффузии и ионная проводимость. Показано, что кооперативная миграция с коллективными смещениями снижает барьеры и важна для прогнозирования свойств легированных литиевых анодов.
-

Работа посвящена созданию автоматизированного подхода к предсказанию адсорбционных свойств металлорганических каркасов с помощью методов активного обучения и молекулярного моделирования, что позволит итеративно исследовать химическое пространство этих структур, упростив поиск наиболее перспективных кандидатов для разделения и хранения газов. Это ускорит открытие новых материалов, способных решать глобальные энергетические и экологические проблемы.
-
В работе рассматриваются молибден-серные нанокластеры как перспективные не платиновые катализаторы реакции выделения водорода (HER) в электролизе воды.
Цель исследования — связать структуру кластеров MonSm с термодинамическими дескрипторами HER и оценить их каталитический потенциал.
-
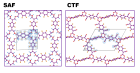
Изучение эксплутационных свойств триазиновых каркасов при помощи методов компьютерного моделирования. Были исследованы следующие свойства: температурная стабильность, оптические, электронные и механические свойства.
-

Работа посвящена разработке методов прогнозирования синтетической доступности материалов по их химическому составу. Для решения задачи использовался подход PU-обучения с использованием предварительно обученного текстового энкодера ModernBERT. Полученные модели демонстрируют высокую точность и могут быть применены для повышения эффективности дизайна новых материалов путем прогнозирования их синтетической доступности.
-

Методами DFT исследована система монослоя MoS₂ с адсорбированной молекулой F4TCNQ. Проанализировано влияние механических деформаций и допирования атомом Br на энергию связывания. Установлено, что растяжение поверхности и наличие Br усиливают адсорбцию.
-

Представлен гибридный подход DFT/ML для оценки устойчивости Tc–C. Сформировано композиционно-конфигурационное пространство углерода в межузельных позициях hcp и fcc Tc (0–20 ат.% C). Интерпретируемые ML-модели выявили соотношения «структура–своиство», а скрининг графовыми неиросетями — наиболее стабильные конфигурации. С учетом вибрационнои свободнои энергии и конфигурационнои энтропии получена фазовая диаграмма 500–2000 K.
-

Методом количественного рекуррентного анализа сигналов акустической эмиссии исследована потеря устойчивости пластической деформации поликристаллов меди и серебра. Показано, что скачок энтропии рекуррентных диаграмм соответствует моменту потери квазистационарности дислокационного ансамбля в модели Кокса–Мекинга. Подход позволяет выявлять предкритические изменения задолго до макроскопического разрушения.