68-я Всероссийская научная конференция МФТИ
Список разделов ФЭФМ - Секция молекулярного моделирования
Заседание секции посвящено докладам студентов и аспирантов, использующих в своих исследованиях методы классической и квантовой молекулярной динамики, метод Монте Карло, методы ab initio расчетов электронной структуры материалов, многомасштабные методы, основанные на атомистическом уровне описания вещества.
Контакты:voronov.iv@phystech.edu
Формат проведения: очный
Дата и время проведения: 04.04.2026 в 10:00
Место проведения: Конференц-зал КСП МФТИ
-
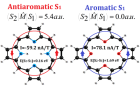
В данной работе исследована возможность применения спектроскопического критерия для быстрой оценки природы магнитно-индуцированных токов в первом возбужденном синглетном состоянии (S1). В качестве модельных систем выбраны ароматический порфин и антиароматический тетраокса-изофлорин. Также были рассмотрены классические представители ароматических соединений: ароматический бензол и антиароматических циклобутадиен, ароматичность S1 состояния которых известна.
-

В данной работе предложен машинно-обучаемый потенциал (МОП) на основе представления окружения через тензоры момента инерции для моделирования зарождения алмазной фазы в разориентированном биграфене. МОП тренировался на наборе структур графена, 2D алмаза и их гидрированных модификаций, полученных расчетами с помощью теории функционала плотности. Обученный МОП воспроизводит энергии и силы этих структур, а также корректно описывает гидрирование двуслойного графена и образование межслойных связей.
-

С использованием крио-ЭМ структур в качестве начального приближения и методов молекулярной динамики было проведено моделирование комплексов mGlu3 с β-аррестином в стехиометриях 2:1 и 2:2. В ходе работы выполнен сравнительный анализ конформационной стабильности, выделены доминирующие конформационные перестройки и определены ключевые участки и аминокислотные остатки, влияющие на связывание β-аррестина.
-
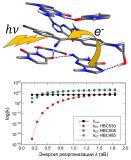
В работе охарактеризованы возбужденные состояния флуорогенов Pepper в изолированном состоянии и в комплексе с РНК. Выделены наиболее вероятные доноры электрона из состава РНК. По теории Маркуса оценены скорости фотоиндуцированного переноса электрона для флуорогенов с высоким и низким квантовым выходом флуоресценции. Показано, что фотоиндуцированный перенос электрона с гуанина G41 на флуороген является возможным каналом безызлучательного распада, снижающим яркость флуоресценции.
-

Представлены два различных распределённых алгоритма метода Хартри-Фока в приближении разложения единицы, реализованных с использованием интерфейсов MPI и OpenMP. Проведено тестирование производительности на суперкомпьютерных вычислительных системах на примере димера молекулы хлорофилла C55H72O5N4Mg в окружении 48 молекул воды. 84- и 71-кратное ускорение достигнуто на 128 потоках. Особое внимание уделено программным оптимизациям алгоритма и перспективам использования реализованных методов.
-

В работе с помощью грубозернистого молекулярного моделирования (силовое поле Martini 3.0, программный пакет GROMACS) исследовано влияние молекулярной структуры неионных ПАВ CnEm на межфазное натяжение на границе вода–алкан. Предложена методика, сочетающая расчёт локального тензора напряжений и реконструкцию поверхности раздела (DBSCAN, триангуляция Делоне), что даёт скорректированные изотермы Гиббса и позволяет определить минимально достижимое натяжение, согласующееся с экспериментом.
-

Значительную долю матриала нефтеносных сланцевых пород составляют керогены - молекулярные предшественники нефти. Рассчёт диффузии в таких пористых материалах требует значительных вычислительных ресурсов ввиду медленных "перескоков" между различными поровыми пространствами. В данной работе были проведены молекулярно динамические рассчёты при помощи которых найдены характерные времена перескоков в матрице керогена и коэффициенты диффузии.
-

Работа посвящена развитию метода расчета матричных элементов спин-орбитального взаимодействия в методе связанных кластеров с многодетерминантным вакуумным состоянием (ic-MRCC). Результаты расчета матричных элементов спин-орбитального взаимодействия для основных состояний атома Si и двухатомной молекулы SeH не демонстрируют принципиально худшей точности при существенно меньшей стоимости вычисления по сравнению с точными аналитическими формулами.
-

В работе исследуется механизм ферментативной реакции гидролиза глутамина в активном центре глутаминазы C с использованием комбинированных методов квантовой и молекулярной механики. Был обнаружен ранее неизвестный реакционный путь, при котором происходит промежуточное ацилирование основной цепи белка.
-

Работа посвящена изучению механизма удлинения ДНК в активном центре ORF2p ретротранспозона LINE-1 без второго катиона магния. С помощью КМ/ММ-моделирования проведена молекулярная динамика, рассчитан профиль энергии Гиббса методом зонтичной выборки и определена эффективная константа скорости (~60 с⁻¹).
-

В работе предложен подход полуколичественной оценки состава сырья для коксообразования по ИК-спектрам. Метод объединяет экспериментальные измерения, молекулярно-динамическое моделирование и хемометрический анализ спектров. Показано, что линейные модели предсказывают с наибольшей точностью массовые доли фракций, тогда как применение нейросетей позволяет улучшить качество предсказания доли CH₃/CH₂. Полученный подход был применен для сопоставления состава сырья с качеством коксования.
-
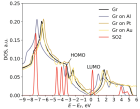
Данная работа посвящена исследованию влияния металлических наночастиц на чувтствительность газовых сенсоров на основе графена. В работе демонстрируется смещение уровня Ферми графена под действием металлической подложки, а также предлагаются механизмы влияния смещения уровня Ферми на частичный перенос заряда между графеном и адсорбируемой на нём молекулой детектируемого газа. Анализ влияния проводится на основе результатов моделирования систем Gr@Me + молекула в рамках DFT.
-

Работа посвящена вопросу возможности модуляции кинетики реакций внешнесферного электронного переноса путём выбора состава электролита. Рассмотрено влияние растворителей диметилсульфоксид и ацетонитрил на распределение компонент редокс-пары ферроцен/ферроцений у поверхности слоистого электрода графен/золото. Показано, что адсорбционные слои растворителей создают высокий потенциальный барьер для проникновения электроактивных частиц непосредственно к поверхности электрода.
-
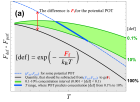
Предлагается метод расчёта энергий образования и миграции точечных дефектов. С помощью предложенного метода рассчитываются коэффициенты самодиффузии в нитриде урана и часть его фазовой диаграммы. Полученные результаты оказываются в разумном согласии с экспериментом
-

Работа посвящена механизмам ферментативного контроля над O₂•⁻, направляющим O₂•⁻-зависимые реакции в продуктивное русло и минимизирующим выход токсичных активных форм кислорода. Методами квантовой химии, КМ/ММ и КМ/ММ МД установлено, что в люциферазе светлячка такой контроль достигается с помощью стерических ограничений в активном центре, кофакторов, подавляющих побочные реакции, и сети водородных связей, обеспечивающей региоселективное присоединение O₂•⁻ к субстрату.
-

Целью работы является создание программного модуля, предоставляющего гибкий и удобный интерфейс для генерации расчетных сеток для численного интегрирования обменно-корреляционных функционалов. Планируется провести оптимизацию модуля с использованием технологий MPI и Kokkos, а также его использование в задаче поиска оптимальных расчетных сеток на различных молекулярных и твердотельных структурах.
-
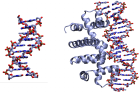
Целью данной работы является сравнительный анализ структурных параметров операторного участка триптофанового репрессора для определения специфичности взаимодействия репрессора с ДНК. Для этого проанализированы данные рентгеноструктурного анализа и из банка данных Protein Data Bank (PDB) последовательности ДНК в свободном состоянии и в составе комплекса с триптофановым репрессором.
-

Квантово-химическое моделирование синтеза аминометоксипроизводных тетрагидрофуранона-3 с бензальдегидом
-

Исследован электронный транспорт через спиральный пиридин-пиррольный олигомер, расположенный между золотыми электродами, с использованием метода DFT+NEGF. Показано, что изменение геометрии спирали и межвиткового взаимодействия приводит к заметной модификации проводимости молекулярного контакта.
-

Предложен байесовский подход к построению фазовых диаграмм и оценке теплоёмкости фторидных систем. Метод сочетает машинное обучение, молекулярную динамику и регрессию гауссовских процессов для восстановления свободной энергии фаз как функции температуры и состава. Теплоёмкость смеси определяется через вторые производные свободной энергии по температуре. Подход применён к бинарной системе NaF–KF и тернарной LiF–NaF–KF, демонстрируя хорошее согласие с экспериментальными данными.
-

В работе представлен байесовский подход к построению фазовых диаграмм бинарных и тернарных систем In–Au–Hg на основе молекулярнодинамических расчетов с машинообучаемым потенциалом MTP. Методология основана на восстановлении свободной энергии Гиббса в зависимости от температуры и концентрации с учетом статистической неопределенности и позволяет выполнять построение фазовых диаграмм и осуществлять прямое сопоставление расчётных и экспериментальных калориметрических зависимостей.
-
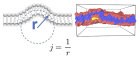
Критическая концентрация белка для начала вытягивания нанотрубок из мембранного бислоя зависит от липидного состава. Известно, например, что присутствие обратных конических липидов ДОФЭ способствует потере устойчивости при меньшем покрытии мембраны белком. Мы построили термодинамическую модель, учитывающую перестановки липидов, которая смогла объяснить этот эффект.
-
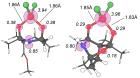
Неинноцентные комплексы могут служить мощным инструментом для придания металлокомплексным катализаторам на основе 3d-металлов реакционной способности, аналогичной реакционной способности систем благородных металлов. В этой работе мы представляем всестороннее компьютерное исследование неинноцентных комплексов, образующихся путем формального внедрения соли железа по связи O–O каркасных органических (амино)пероксидов.
-

В работе реализован автоматизированный расчет индекса пластичности ОЦК-сплавов с использованием машиннообучаемого потенциала MTP.