67-я Всероссийская научная конференция МФТИ
Список разделов ФЭФМ - Секция молекулярного моделирования
Заседание секции посвящено докладам студентов и аспирантов, использующих в своих исследованиях методы классической и квантовой молекулярной динамики, метод Монте Карло, методы ab initio расчетов электронной структуры материалов, многомасштабные методы, основанные на атомистическом уровне описания вещества.
Формат проведения: очный
Дата и время проведения: 04.04.2025 в 10.00
Место проведения: МФТИ, Конференц-зал КСП (2-й этаж)
-

Дихалькогениды переходных металлов (MoS2), привлекают внимание благодаря способности к фазовым переходам между полупроводниковыми (2H) и металлическими (1T, 1T’) фазами. В данном исследовании с использованием DFT предсказаны новые металлические фазы (1T’ и 1T’’) для нанолент Ta₂Pd₃Se₈ и Ta₂Ni₃Se₈, изучены их электронные свойства и энергетические барьеры переходов.
-

В работе рассматриваются формы раздела поверхностей жидкость-жидкость в узких порах при периодических граничных условиях. Для каждой конфигурации поверхности выводятся аналитические выражения для профилей плотности, площади поверхности и условий существования.
-

Теоретическая работа, проведённая на базе эксперимента, посвящена изучению влияния электрического поля на стабильность наноструктуры биграфен/диаман, демонстрирующую явление резистивного переключения. с использованием метода теории функционала плотности (DFT) были объяснены механизмы, обеспечивающие уникальные свойства данной структуры.
-
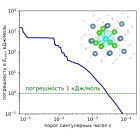
В настоящей работе рассматривается эффективность использования тензорного разложения Таккера для снижения вычислительной сложности релятивистского метода связанных кластеров, учитывающего одно- и двукратные возбуждения (модель CCSD).
-

В работе рассматриваются различные подходы к реализации алгоритма метода Хартри-Фока в приближении разложения единицы с помощью параллельных вычислительных технологий.
-

Целью данной работы являлось сравнение строения и каталитической активности трех активных центров F1-ATPазы. В работе провели расчеты молекулярной динамики с потенциалами комбинированного метода квантовой механики / молекулярной механики (КМ/ММ) в течении 10 пс для анализа строения активных центров в фермент-субстратном комплексе.
-

Недавно было показано, что биграфен, повернутый на 30 градусов, образует многосвязный граф, позволяющий построить муаровый квазиузор. Анализ продемонстрировал самоподобие внутри этих фигур при масштабировании узора на определенный коэффициент. В данной работе проведено построение данного узора и его масштабирование на квадрат константы решетки, далее методом Монте-Карло с использованием потенциала машинного обучения проведено атомистическое моделирование осаждения водорода на биграфен.
-

На первом этапе работы было проведено моделирование облучения многослойного графена высокоэнергетичным ионом Xe. Затем предложены модели структур, которые могут частично описать наблюдаемый фазовый переход. Далее с помощью метода TDDFT рассмотрены одноэлектронные возбуждения в изучаемых углеродных кластерах со смешанными типами связей sp2-sp3.
-

Рассчитано линейное натяжение дислокации в диоксиде урана при помощи атомистического моделирования. Предложена теоретическая оценка условия отрыва дислокации от пузырька и оценена максимальная сила взаимодействия дислокации с пузырьком.
-
Структуры электролит/графен/металл интересны с фундаментальной и прикладной точек зрения из-за возможности сильно изменять свойства графена вследствие его низкой плотности электронных состояний в окрестности уровня Ферми. В данной работе исследованы свойства межфазных границ электролит/графен и графен/золото с точки зрения образования эффективных конденсаторов на контакте фаз и возникновения дополнительных источников скачка потенциала при изменении потенциала поверхности.
-
Работа выполнена в рамках задачи о повышении чувствительности и селективности газовых сенсоров на основе графена. В работе было проведено моделирование электронной структуры систем метал-графен-молекула в рамках DFT для металов: алюминий, платина; молекул газа: оксид серы, аммиак; а также при наличии гидроксильной группы на графене и без неё.
-

Учитывая разнообразие флуоресцентных белков при неизменной структуре хромофора, обусловленное эффектом Штарковской настройки, мы предлагаемый оригинальный подход, предполагающий построение общей предсказательной модели машинного обучения, обеспечивающей физически интерпретируемое предсказание спектров флуоресцентных белков в рамках концепции «структура-свойство».
-

В данной работе проведены вычислительные эксперименты методом классической молекулярной динамики для исследования сдвиговой вязкости водного раствора хлорида натрия. Анализ учитывает влияние моляльной концентрации ионов Na-Cl, температуры системы и зарядов погруженных ионов. Полученные результаты показали, что силовые параметры погруженных ионов существенно влияют на вязкостные характеристики раствора, что важно для моделирования транспортных свойств электролитов в различных условиях.
-

В работе представлен расчет энергетических уровней атома кремния методом связанных кластеров с многодетерминантным вакуумом (ic-MRCC) реализованный в открытом программном обеспечении GeCCo в связке с квантовохимическим пакетом PySCF, а также разработанный интерфейс pyscf2gecco для передачи молекулярных интегралов в правильном формате между программами.
-

Учет межмолекулярной электронной корреляция является необходимым требованием для описания межмолекулярных взаимодействий. Применение явно коррелированного подхода требует больших расчетных мощностей, поэтому в качестве альтернативы может использоваться дополнительный набор связевых функций в сочетании с высокоточным однореференсным квантово-химическим методом, как метод связанных кластеров CCSD(T).
-
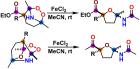
Проведено квантово-химическое исследование трансформации каркасных аминопероксидов под действием FeCl₂, приводящее к образованию замещённых тетрагидрофуранов через каскад разрывов связей O–O и C–C. Анализ показал, что радикальный характер кислородных лигандов и стабилизирующая роль железа обеспечивают эффективность реакции.
-

В данной работе представлены результаты исследования взаимосвязи геометрии активного центра миозина и поведения лапласиана электронной плотности в критической точке связи
-

Работа посвящена изучению механизма направленного действия оксирановой группы лиганда в качестве ингибитора на остатки цистеина и аспартата в онкобелках K-Ras(G12C) и K-Ras(G12D) соответственно с помощью молекулярной динамики с потенциалами комбинированного метода квантовой механики/молекулярной механики (КМ/ММ): для геометрических оценок – без добавления смещающего потенциала, для построения профиля – с добавлением смещающего потенциала методом зонтичной выборки.
-

В настоящей работе для моделирования системы был написан код на языке Пайтон, включающий расчет энергии системы в приближении парных потенциалов, а также расчет функций ядерных градиентов и компонент гессиана. Была проведена оптимизация геометрии кластера с использованием градиентных методов, исследована его стабильность, и проведен анализ точности и устойчивости решений с использованием методов классической молекулярной динамики
-
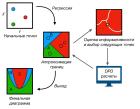
В данной работе в рамках крупнозернистого компьютерного моделирования разработана оптимизационная схема направленного поиска параметров сополимеров, при которых наблюдается самоорганизация мембранных структур (везикул) в растворе.
-
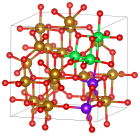
Предложен способ расчета концентраций точечных дефектов в железохромистых шпинелях в зависимости от температуры и давления кислорода, основанный на результатах расчетов в рамках DFT+U и экспериментальных данных
-

В данной работе с использованием метода DFT и модели сольватации IEFPCM анализируется влияние различных растворителей на стабильность и геометрию стэкинговых комплексов.